[Share] Some thoughts on NIST's development of ERBB2-CNV standard
Generally speaking, copy number variant (CNV) is defined as the amplification or reduction of DNA fragments of 1kb or larger in the human genome. CNVs can be divided into copy number gain (copy number gain) and copy number deletion ( copy number loss), in tumor cells, the amplification of proto-oncogenes and the deletion of tumor suppressor genes often occur. The amplification and deletion generally directly affect the expression of RNA and protein, which in turn affects the occurrence and development of tumors. Cell growth, drug sensitivity, and drug resistance are closely related. There are data showing that CNVs are ubiquitous in the human cancer genome, affecting larger segments of the genome than single nucleotide polymorphisms (SNPs).
There are various methods for the detection of gene CNV, including Southern blot hybridization, fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), comparative genome hybridization (CGH), whole genome single nucleotide polymorphism (SNP) array And NGS, etc., each has advantages and disadvantages.
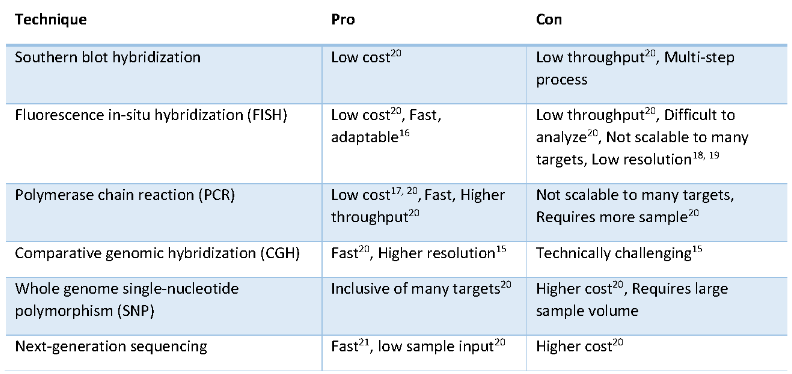
Fig 1. CNV检测技术比较
Today we share with you a NIST(National Institute of Standards and Technology)The article (click to view the content of the article) is their process of developing 2373 reference materials: gDNA standards for HER2 are mainly used for Q-PCR and dPCR, and of course for NGS, but due to different detection methods, Different analysis methods will be inconsistent with Q-PCR and dPCR in quantitative values.
First of all, the article found that many samples of breast cancer patients have a large number of chromosomal structural abnormalities. When developing Q-PCR and dPCR, it is necessary to select internal reference genes, and the selection of internal reference genes should try to avoid chromosomal abnormal regions. The study found that these breast cancer samples often have gene amplification in 1q, 8q, 20q, 7, 11q, 13, 17q, 9q, 16p and other regions, and gene amplification frequently occurs in 8p, 11q14-qter, 18q, Xq and other regions. At the same time, the selection of the internal reference gene should not be selected on chr17 (the location of the HER2 gene), so the article finally selected the DCK gene, EIF5B gene, RPS27A gene and PMM1 gene (PMM1 is only used for Q-PCR experiments).

Table 1. Primer information for HER2 and reference genes
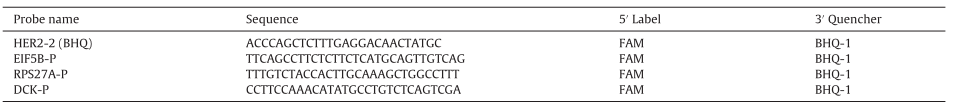
Table 2. Probe information for HER2 and reference genes
As a standard, it is necessary to simulate clinical samples as much as possible, and at the same time, a stable long-term supply is required. Therefore, the article selects 5 common breast cancer cells as the raw materials of the standard for calibration experiments, namely: SK-BR-3 ( components A), MDA-MB-231(components B), MDA-MB-361(components C), MDA-MB-453(components D) and BT-474(components E), these cells were cultured, gDNA was extracted, STR identification, to ensure qualified quality, ready for the next test.
At the same time, the article constructed a HER2 cDNA plasmid, calculated the corresponding relationship between mass and copy number through the molecular weight of the plasmid, and constructed a standard curve. At the same time, the 2372-A sample (whole genome DNA: gDNA) was introduced, and the mass and copy number were also constructed. Through Q-PCR detection, it was found that there was a strong correlation, R2=0.9924, and the 2372-A sample was diploid, and the corresponding HER2 copy number was 2, which could be used for the detection and calibration of components AE.
Next, Q-PCR and dPCR were used to detect components AE. Four internal reference genes were used for Q-PCR, and the copies/μL of AE samples were calculated according to the calibration curve. Three internal reference genes were used for dPCR. The ratio of copies/μL and internal reference genes, here, two dPCR methods were used in the article, namely droplet dPCR and chamber dPCR.
In terms of calculation, first, the 2372-A gDNA sample was corrected by the Q-PCR data of the plasmid HER2 cDNA to show that the 2372-A sample was a diploid sample with 2 copies of HER2, and then the 2372-A sample was used to calculate the copy number of the AE sample. (copies/μL), dPCR directly detects the copy number of HER2 and the internal reference gene, and then calculates the ratio.
The final article showed that the first two dPCRs used three internal reference genes to calculate the ratio of HER2 to the internal reference gene. The results were very consistent. The five samples showed varying degrees of copy number amplification, ranging from weak to strong.
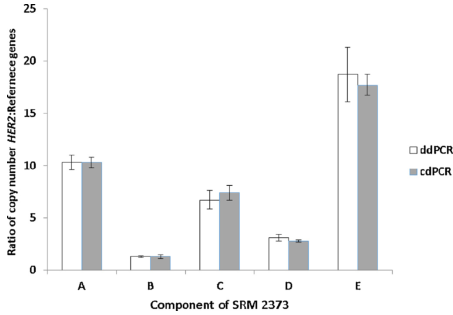
Fig 2. For A-E samples, the two dPCR methods were consistent. (The data is the average of 3 internal parameters)
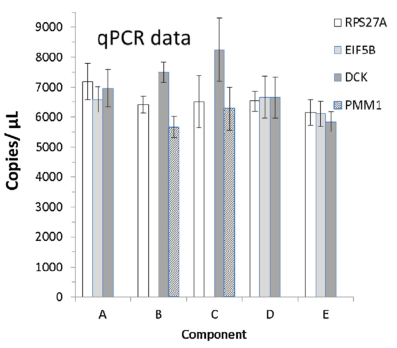
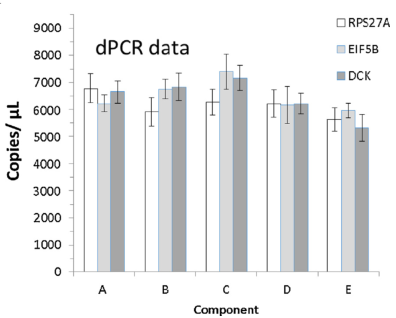
Secondly, A-E samples also showed strong consistency for copies/μL in Q-PCR and dPCR, indicating that A-E standards are suitable for both Q-PCR and dPCR applications.
|
|
Fig 3. The left side is the copy value of the internal control in the Q-PCR detection of the A-E samples, and the right side is the copy value of the internal control in the A-E sample in the dPCR detection. | |
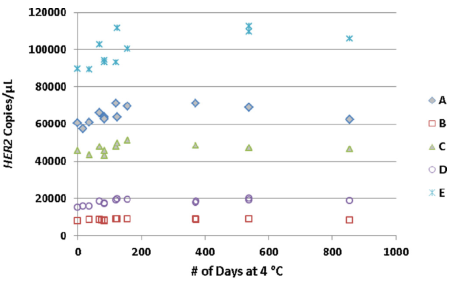
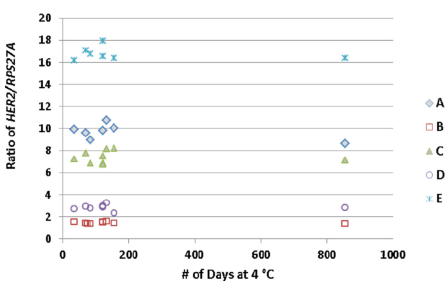
In addition, the article also tested the stability of A-E samples. In the 856-day test, both the absolute copy number and the ratio to the internal reference showed good stability.
|
|
Fig 4. Both the copy number of HER2 and the ratio to the internal reference were relatively stable in the 856-day test. | |
Finally, the article also tested the effect of random factors on the repeatability of A-E experiments, such as machine, operator, date, etc., showing good repeatability.
In summary, when developing molecular diagnostic standards for CNV detection, we need to pay attention to:
● The absolute copy number (copies/μL) can be set as a standard, and dPCR and Q-PCR calibration can be used, where Q-PCR needs to use a calibration standard;
● The ratio to the reference gene can be set. As a standard, it is suitable for the method of dPCR (Q-PCR can also be used, and the calibration curve is also required). At this time, it is necessary to pay attention to the selection principle of the internal reference. If possible, try to select as many internal reference genes as possible;
● Standard products need stable supply, long-term stability needs to be tested, and repeatability under different random factors needs to be tested;
However, we found that there are still some problems that have not been solved, such as: Q-PCR and dPCR are PCR reactions of very short sequences. Which segment of the entire HER2 gene is selected for amplification? The amplification efficiency of different segments will directly affect the copy number (copies/μL), and then the ratio of the copy number detected by different segments to the internal reference gene will be inconsistent, and even whether the PCR reaction is just designed in each copy. The difference is not just a matter of amplification efficiency.
In addition to the article, we can explore: Is this standard suitable for NGS? We know that NGS compares the sequencing depth of various places, and then judges the copy number of the region. It is very different from the detection methods of Q-PCR and dPCR. It seems that NGS will detect different results.

